|
|
- The function of the menu boxes / visual elements - click on the menu box / visual element of interest.

- Parameter names and values - this panel shows the names and values of the fit parameters as well as the subspectrum headlines associated with the selected fit model, along with visual elements like the navigation / menu shapes (square or circle) before the parameter names, and the value-setting bars after the parameter values. In connection with the subspectrum headlines and the parameter names and values the following functionalities are available.
- in order to edit the value in question. During editing characters can be selected by mouse, and selected (white) characters can be copied into the clipboard by pressing ctrl+C, as well as they can be pasted from the clipboard at the cursor position by pressing ctrl+V. In order to finish editing press enter or click with the mouse outside of the editing area. In order to cancel editing, press ESC.
- in order to fix / unfix the parameter in question. An unshared parameter is fixed if it is displayed with red color and it is free if it is displayed with darkgray color. The value of fixed parameters do not change during fitting.
- in order to fix / unfix the parameter in question. A shared parameter can be fixed either independent of or together with the corresponding parameters in other - simultaneously fitted - spectra (example). Independently fixed shared parameters are displayed with red color. Shared parameters that are fixed to the same value in all the corresponding shared subspectra (sharefixed parameters - as we prefer to call them) are displayed by magenta color.
A shared parameter is displayed with blue color if it is unfixed. The value of sharefixed parameters do not change during fitting.
- in order to share / unshare the parameter in question. A shared parameter can be unshared either by detaching it from the rest of the corresponding shared parameters (which latter will remain shared), or by setting the status of all the corresponding parameters to unshared / independent (example).
- A parameter is unshared if its value influences only the fit model set for the current spectrum.
- A parameter is shared if it is enforced to have the same value as the corresponding parameters in the models set for other spectra, and vice versa. A parameter can be defined to be shared only if simultaneous fitting of two or more spectra is performed. Only shared subspectra can have shared parameters.
- A subspectrum is shared if it exists in the model of two or more - simultaneously fitted - spectra. A fit model can include shared as well as unshared subspectra at the same time. The navigation / menu shapes of shared subspectra are circles (example). A shared subspectrum can be added to the fit model via the ADD menu box. The physical model associated with a shared subspectrum can only be changed coherently among the corresponding shared subspectra, i.e. if a shared subspectrum is modified in one of the simultaneously fitted spectra, then the modifications will take place in other simultaneously fitted spectra as well.
- A subspectrum is unshared if it is independent of the subspectra being part of models set for other - simultaneously fitted - spectra. Unshared subspectra cannot have shared parameters. The navigation / menu shapes of unshared subspectra are squares (example).
- in order to access the subspectrum's headline menu list with the following options.
- Rename subspectrum
- Add one more subspectrum of this type
- Unshare subspectrum in all of the spectra
- Unshare subspectrum in this spectrum only
- Delete subspectrum
- Fix all subspectrum parameters
- Unfix all subspectrum parameters
- Constrain parameters to be equal to those of
- Fit only this subspectrum
- Fit all but this subspectrum
- Copy peak positions and areas to clipboard / including information on nuclear states (example)
- Navigation / menu shapes (square or circle) help navigation among the parameters as they are displayed in the model page, as well as provide access to a popup menu with options concerning the corresponding subspectrum.
Move the mouse pointer over the shape before a parameter in order to achieve the selection of the parameter on the model page. Press on one of the shapes in order to access the headline menu list of the corresponding subspectrum.
The navigation / menu shapes of unshared subspectra are squares, while those of shared subspectra are circles.
Parameters whose meaning/influence has been changed on the details page (with respect to their default meaning/influence for the given subspectrum type) are denoted by a menu shape with a light yellow center (example).
Shared parameters for which the sharing constraint could not be satisfied for one or more of the simultaneously fitted spectra are denoted with a "strikethrough circle" (see below).
 - Parameter of an unshared subspectrum. - Parameter of an unshared subspectrum. - Parameter of an unshared subspectrum describing a linear model that was modified by the user such that the meaning of the parameter is unlike its default meaning. (The differences can be checked on the details page.) - Parameter of an unshared subspectrum describing a linear model that was modified by the user such that the meaning of the parameter is unlike its default meaning. (The differences can be checked on the details page.) - Parameter of a shared subspectrum. - Parameter of a shared subspectrum. - Parameter of a shared subspectrum describing a linear model that was modified by the user such that the meaning of the parameter is unlike its default meaning. (The differences can be checked on the details page.) - Parameter of a shared subspectrum describing a linear model that was modified by the user such that the meaning of the parameter is unlike its default meaning. (The differences can be checked on the details page.) - Parameter of a shared subspectrum for which a sharing constraint was set up, but for one or more of the simultaneously fitted spectra the sharing constraint could not be satisfied. (This may occur, e.g., for shared percentage parameters that may be subject to conflicting constraints in the different spectra.) - Parameter of a shared subspectrum for which a sharing constraint was set up, but for one or more of the simultaneously fitted spectra the sharing constraint could not be satisfied. (This may occur, e.g., for shared percentage parameters that may be subject to conflicting constraints in the different spectra.)
- Value-setting bars show the relative position of the corresponding parameter's current value between its allowed minimum and maximum values.
If the current value of the parameter is lower than its minimum value, then the bar changes to a green arrow, whereas if it is higher than the allowed maximum value, then the bar changes to a red arrow (example).
Press on the bar with the left mouse button in order to change the corresponding parameter value.
In order to change the minimum or the maximum value allowed for a parameter, while pressing the left mouse button, move the mouse pointer outside the left- or right-side limit of the bar, and then press the right mouse button in addition to the left one.
- Down / Up - Press on these boxes in order to scroll the parameter list such that parameters below / above the lower / upper edge of the parameters' panel become visible and editable. Alternatively, the down / up arrow keys, the Page Down / Page Up keys, as well as the mouse wheel can also be used to achieve the same effect.
- The goodness of the (simultaneous) fit is emphasized in the bottom of the parameters' panel in the form of
Chi: () []
If the chosen theoretical model is correct, then an acceptable fit should provide a normalized chisquare close to 1, and a goodness of fit value higher than about 0.001 .
In the case of simultaneous fitting of several spectra, these values characterize the simultaneous fit as a whole. In contrast, the corresponding values visible on the spectrum panel characterize only the fit of the current spectrum, as if it was the only one that is fitted. Accordingly, as long as only a single spectrum is fitted, the two panels show identical goodness-of-fit values.
- Group - Press on this menu box in order to change the apparent order of the subspectrum fit parameters (i.e. those following the parameter group of the base line) by selecting one of the following options.
- - Select this option in order to have fit parameters grouped first by subspectra and then by the type of parameters. Parameters belonging to the same subspectrum will follow each other in the following order: first the amplitude type parameters, then the position type parameters (which influence the position of the absorption peaks), then the width type parameters, and then the extra parameters if any. Each subspectrum parameter group is introduced by a headline showing the name and relative area fraction (in percentage) of the subspectrum, as well as a read-only bar whose length is proportional to the shown percentage value (example).
- - Select this option in order to have fit parameters grouped first by the type of parameters and then by the subspectra they belong to. First the amplitude-, then the position-, then the width- and finally the extra type parameters — if any — will be displayed. Parameters of the same type will be grouped by subspectra (example).
- - Select this option in order to have fit parameters grouped by the type of parameters and to have parameters of the same type ordered alphabetically. First the amplitude-, then the position-, then the width- and finally the extra type parameters — if any — will be displayed in alphabetical order (example).
- - Select this option in order to show / hide subspectrum headlines when fit parameters are grouped first by subspectra.
- S k/n indicates that the current spectrum is the kth from the n simultaneously fitted spectra. Press the TAB or the Backspace keys on the keyboard in order to navigate between the different simultaneously fitted spectra. If only a single spectrum is fitted, then S 1/1 is displayed.
When simultaneous fitting of several spectra is performed, press on the corresponding box in order to access the following menu options in the appearing popup.
- - select to order the fitted spectra alphabetically according to their file name.
- - select a fit parameter from the appearing list to have spectra ordered in an ascending manner with respect to the value of the selected parameter.
- - select a special parameter from the appearing list to have spectra ordered in an ascending manner with respect to the value of the selected parameter.
- - select this option to reset the original order of spectra that was set automatically when the FIT menu was entered.
The spectrum order set up via the above options will influence the order in which the spectra will follow each other when the TAB and Backspace keys are pressed,
the order in which the spectra follow each other on the All Spectra tab, as well as the order in which spectra will appear on images printed to printer or copied to the clipboard.
- P n - indicates that the number of free parameters is equal to n. Free parameters are those which are actually adjusted / fitted when a fit procedure is initiated.
Press on the corresponding box in order to access the following menu options in the appearing popup.
- - Select this option in order to achieve the following changes in the fix status of the fit parameters of the current spectrum:
- Free → Fixed
- Shared → Independently fixed shared
- - Select this option in order to achieve the following changes in the fix status of the fit parameters of the current spectrum:
- Fixed → Free
- Independently fixed shared → Shared
- Independently fixed sharefixed → Sharefixed
- - Select this option in order to achieve the following changes in the fix status of the fit parameters of the current spectrum:
- Fixed ↔ Free
- Sharefixed ↔ Shared
- Independently fixed sharefixed ↔ Independently fixed shared
- - Select this option in order to achieve the following changes in the fix status of all the fit parameters:
- Free → Fixed
- Shared → Sharefixed
- Independently fixed shared → Independently fixed sharefixed
- - Select this option in order to achieve the following changes in the fix status of all the fit parameters:
- Fixed → Free
- Sharefixed → Shared
- Independently fixed shared → Shared
- Independently fixed sharefixed → Shared
- - Select this option in order to achieve the following changes in the fix status of all the fit parameters:
- Fixed ↔ Free
- Sharefixed ↔ Shared
- Independently fixed sharefixed ↔ Independently fixed shared
(The fix status of nuclear and user-defined extra parameters — if any — will not be altered by selecting the above options.)
- Bars - Press on this tab in order to have the value-setting bars displayed on the right-hand side of the parameters' panel.
- StD - Press on this tab in order to follow the changes in the parameter values during fitting, or to display the standard deviation values of the parameters (example) after the StD values were calculated via a press on the Cal StD box.
During fitting the displayed deviation values are calculated by subtracting the parameter's previous value from its new value, i.e. if the deviation is positive, then the parameter has increased in the last step of the fit. In order to change between the value-setting bars and the deviation values during fitting, position the mouse pointer over the parameters' panel, and then press space on the keyboard.
- Chi - Press on this box in order to access the following options related to the fitness function and the corresponding calculation of the goodness-of-fit parameter.
- - Select this option to have chisquare minimized during fitting.
- - Select this option to have the sum of simple squared deviations - between the measured and calculated data points - minimized during fitting.
- - Select this option to have the sum of absolute deviations - between the measured and calculated data points - minimized during fitting.
- - Select this option in order to have zero-valued data points of the current spectrum considered during fitting.
- - Select this option in order to have zero-valued data points ignored in all the fitted spectra. Ignored data points are not taken into account when the goodness of the fit is calculated, consequently they do not influence the results of the fit.
- LinFit - Press on the LinFit box with the left mouse button in order to optimize the Base Line and Amplitude type parameters of the model set for the current spectrum in a single step.
Press on it with the right mouse button in order to do the same optimization for all the simultaneously fitted spectra. The value of position-, width- and extra type parameters will not be altered.
- Amplitude / Area - Press on this box in order to toggle between Amplitude and Area modes of the calculation of individual absorption peaks. This setting influences all the simultaneously fitted spectra.
When area mode is selected then amplitude type parameters are treated as area type parameters, i.e. they are assumed to determine the area under individual peaks. If amplitude mode is selected, then the amplitude-type parameters determine the amplitude of the individual peaks. At the same time, even if area / amplitude mode is selected, amplitude-type parameters are not necessarily equal to the area / amplitude of the associated peaks, because the factors connecting the peak parameters to the amplitude-type fit parameters may also influence the calculated peak areas / amplitudes. (These factors can be set on the Details page).
Independent of whether Amplitude or Area mode is set, for theoretical models that cannot be altered by such factors (e.g. Hamiltonian models), the amplitude-type parameter always denotes the area under the whole subspectrum.
The area under the individual subspectra can also be calculated on the basis of the Total Spectrum Area (TSA) and the relative subspectrum areas displayed on the fit report for each of the subspectra being part of the fitted model.
- Transmission / Reflection - Press on this box in order to toggle between transmission and reflection geometry modes for the current spectrum. Transmission refers to transmission Mossbauer spectroscopy where peaks are subtracted from the background, whereas Reflection refers to scattering geometries like CEMS or XMS where the peaks are added to the background.
- Exit - Press on this box with the left mouse button to leave the FIT menu and have distribution curves — if any — appearing in the main menu as separate windows. Press on it with the right mouse button in order to leave the fit menu without having distribution curves to appear in the main menu.
Note that fit results are not automatically saved to the fitted spectra - press on the Accept box in order to achieve that.
- Global - Press on the Global menu box in order to initiate global fitting of model parameters. The global fitting procedure is based on a robust numerical optimization method realized by a special evolution algorithm developed especially for the purposes of Mossbauer spectrum analysis.
(See NIM B 129 (1997) 527 for details.) Global fitting is able to locate the global optimum of the set of fitting parameters even if the initial parameter set is very far from being optimal. Global fitting fully respects the allowed parameter ranges, i.e. it will never produce a result that is outside of these ranges. (Consequently, if the optimum solution is outside of the preset ranges, then the global fitting procedure cannot succeed.)
Global fitting is infinite: press the ESC key on the keyboard in order to terminate the fit. After global fitting was stopped, one usually turns to the FIT menu box in order to finish up the fit by local tuning of the fit parameters.
- FIT - Press on the FIT menu box with the left mouse button in order to initiate local tuning of the fit parameters starting from the current parameter set. Local tuning does not respect the constraints imposed on the allowed parameter ranges, unless the constraints in question are designated to be HARD. Local tuning stops automatically after it has been converged with high precision. However, if lower precision is also acceptable one can also stop the fit manually by pressing the ESC key on the keyboard.
Whether manual stopping resulted in an acceptable solution can be verified by calculating the standard deviation of the parameters via the Cal StD menu box: if the standard deviation values can be calculated without MossWinn reporting about an error, then the manual stopping of the fit procedure was valid.
Press on the FIT menu box with the right mouse button in order to access one of the following options.
- - Initiates local tuning of the fit parameters by using Method A (equivalent to the left mouse click on the FIT menu box).
- - Initiates local tuning of the fit parameters by using Method B that is more robust but - on average - converges slower than Method A.
- - Select this option to optimize the Base Line and Amplitude type parameters of the model set for the current spectrum in a single step (eqivalent to the effect of a left mouse click on the LinFit box).
- - Select this option in order to adjust only those fit parameters that are part of the model set for the current spectrum. It is the simultaneous goodness of fit that is optimized, such that in the case of simultaneous fitting of multiple spectra - and in the presence of shared parameters - the final result may also be influenced by the other spectra fitted.
- - Select this option in order to carry out the previous function (Fit current spectrum only) for all the simultaneously fitted spectra one after the other. After the last spectrum was also fitted in this way, the fit starts again with the first one, and so on until convergency is achieved.
- - Select this option in order to fit only the base line and the amplitude-type parameters for all the simultaneously fitted spectra.
- - Select this option in order to fit only the position-type parameters for all the simultaneously fitted spectra.
- - Select this option in order to fit only the width-type parameters for all the simultaneously fitted spectra.
- - Select this option in order to fit only the extra-type parameters (i.e. those which follow the width-type parameters) for all the simultaneously fitted spectra.
- Cal StD - Press on the Cal StD menu box with the left mouse button in order to calculate the standard deviation of the fitting parameters by assuming that they represent the result of a convergent fit. The calculations are based on the approximation of the curvature matrix (one-half times the Hessian matrix) of the chisquare quantity. The method used for the calculation of the chisquare quantity depends on the fitness function optimized during the fit.
The calculations can proceed via the numerical approximation of either the 1st derivatives of the fit model envelope or the 2nd derivatives of the chisquare quantity with respect to the fit parameters. Turn to the Configuration settings... option of the SET menu, in order to determine which of the two methods gets invoked by pressing on the Cal StD menu box.
Press on the Cal StD menu box with the right mouse button in order to access one of the following options.
- - Turn to this option in order to copy the correlation matrix of fit parameters into the Windows clipboard. Element (i,j) of the correlation matrix is calculated as COV(i,j)/[COV(i,i)COV(j,j)]½ where COV(i,j) denotes element (i,j) of the covariance matrix, the latter being equal to the inverse of the curvature matrix of chisquare.
The correlation matrix becomes recalculated each time the StD of parameters is calculated via the inversion of the curvature matrix. As the output format of the correlation matrix the following options are available.
- As image (for all spectra) - The correlation matrix will be copied into the clipboard as a grayscale bitmap image. All the fitted spectra are considered. The cells of the matrix image are shaded according to the corresponding correlation value: higher correlation values are denoted by a darker shade of gray. Spectra, subspectra and parameters are shaded as their darkest-shaded subspectrum, parameter and correlation value, respectively (example). Invalid values (either referring to a fit that is not optimal, or signalling the occurrence of a singularity during matrix inversion) are denoted by a question mark on a black background.
- As image (for current spectrum) - The correlation matrix will be copied into the clipboard as a grayscale bitmap image. Only those fit parameters are considered, that belong to the fit model of the current spectrum.
- As image (value limited, for all spectra) - The correlation matrix will be copied into the clipboard as a grayscale bitmap image. Only those fit parameters are considered that display correlation values higher than or equal to the value set by the user. This option is especially useful to reduce the size of the correlation matrix - and restrict it to the potentially most problematic parameters - by excluding parameters that do not display appreciable correlation.
- As text (for all spectra) - The correlation matrix will be copied into the clipboard as a table given as comma delimited text. All the fitted spectra are considered.
- As text (for current spectrum) - The correlation matrix will be copied into the clipboard as a table given as comma delimited text. Only those fit parameters are considered, that belong to the fit model of the current spectrum.
- As text (value limited, for all spectra) - The correlation matrix will be copied into the clipboard as a table given as comma delimited text. Only those fit parameters are considered that display correlation values higher than or equal to the value set by the user. This option is especially useful to reduce the size of the correlation matrix - and restrict it to the potentially most problematic parameters - by excluding parameters that do not display appreciable correlation.
- - Turn to this option in order to save the correlation matrix of fit parameters as a GIF image file that is optimal for being inspected by the help of web browsers. As the output format of the correlation matrix the following options are available.
- As GIF image (for all spectra) - The GIF image of the correlation matrix is saved by considering all the fitted spectra.
- As GIF image (for current spectrum) - The GIF image of the correlation matrix is saved by considering only those fit parameters that belong to the fit model of the current spectrum.
- As GIF image (value limited, for all spectra) - The GIF image of the correlation matrix is saved by considering only those fit parameters that display correlation values higher than or equal to the value set by the user.
- - Select this option in order to calculate the standard deviation of the fit parameters via the approximation of the curvature matrix on the basis of the numerical approximation of the 1st derivatives of the fit model envelope with respect to the fit parameters, by assuming that the current set of fit parameters represents the result of a convergent fit.
This method is in general faster and more reliable — especially in cases involving the fit of hyperfine parameter distributions — than the method based on the approximation of the chisquare 2nd derivatives.
- - Select this option in order to calculate the standard deviation of the fit parameters via the approximation of the curvature matrix on the basis of the numerical approximation of the 2nd derivatives of the chisquare quantity with respect to the fit parameters, by assuming that the current set of fit parameters represents the result of a convergent fit.
This method is susceptible to failure when the fit involves hyperfine parameter distributions, in which case the method based on the approximation of the 1st derivatives could be used to observe the StD values of fit parameters.
- - Select this option in order to calculate the standard deviation of the fit parameters via Monte Carlo iterations by assuming that the current set of fit parameters represents the result of a convergent fit.
This method is more robust but works slower than the default method based on the approximation of the curvature matrix. Assuming that the fitting envelop represents well the measured data, and that the measured counts distribute around their expected values normally,
the program generates hypothetical spectra (that could have been observed in repeated measurements) and fits the theoretical model to all of them one after the other. (When the fitness function is the the variance of the counts is assumed to be equal to the value of the counts in accordance with the underlying Poisson statistics, whereas when the fitness function is the or the , a uniform variance is assumed and estimated on the basis of the counts and the fit model curve.)
The standard deviation of the model parameters is then estimated on the basis of the statistics of the parameter values obtained in the subsequent fits.
100 iterations usually provide sufficient precision in practice. By the use of this method one can also estimate the reliability of distribution data points: the program records the value of every distribution data point during the subsequent iterations, and on the distribution graph it draws the minimum and the maximum for each data point by blue and red color, respectively.
On the basis of the figure observed (example) in this way one can have an impression of how reliable the observed distribution is.
- - Select this option in order to clear the calculated standard deviation values of the fit parameters.
- - Based on the principle of Monte Carlo iterations detailed above, this option provides a faster alternative for the estimation of the reliability of distributions.
In the case of the present option the fit parameters are kept fixed to their original values, and the fit of the hypothetical spectra is optimized only by recalculating the distribution curve. Although this method can be expected to show distribution curves to be more reliable than they actually are, in practice it is a rather useful method especially to rule out unreliable distribution curves.
- - Turn to this option in order to remove the blue (minimum) and red (maximum) curves - calculated by the above mentioned Monte Carlo error estimation procedures - from the distribution graphs (if any).
- Accept - Press on the Accept menu box with the left mouse button in order to save the current status of the fit to the data file associated with the fitted spectrum. The fit result accepted in this way becomes automatically reloaded when next time the FIT menu is entered for the same spectrum.
Accepting a fit result will overwrite any fit result accepted previously for the same spectrum file.
In the case of simultaneous fitting of several spectra, the complete status of the fit is saved, and the simultaneous fit is recovered when next time the fit menu is entered for any of the spectra for which the corresponding simultaneous fit result was accepted. (To accomplish this recovery task, MossWinn may need to load in spectra if required by the saved simultaneous fit, which is then carried out automatically while the FIT menu is being entered.)
Normally, after a successful fit was carried out, it is recommended to accept the fit. (Note that the TBL menu creates tables of fit parameters on the basis of accepted fit results.)
When the option of is enabled via the performance settings, then the current status of the fit is furthermore also saved (cumulatively, but without duplicates) to the respective HTML FitLog files of the fitted spectra.
In order to access further functions related to the accepted / logged fit statuses, press on the Accept menu box with the right mouse button, and select one of the following options.
- - Select this option in order to reload the value of those actually defined fit parameters that were also included in the fit accepted previously for the current spectrum (via an LMC on the Accept menu box). (The fit model itself is not reloaded, i.e. the number and kind of the subspectra — contributing to the actually set fit model — will not be altered.)
- - Select this option in order to save a HTML FitLog to the FitLog file of the current spectrum, according to the current state of the fit.
- - Select one of the submenus of this menu option in order to reload and set the corresponding fit model from the FitLog file associated with the current spectrum. The currently set fit model will be overwritten.
- - Select one of the submenus of this menu option in order to remove the corresponding FitLog from the FitLog file associated with the current spectrum.
- - Select this menu option in order to remove all but the last saved FitLog from the FitLog file associated with the current spectrum. Select the appearing submenu item in order to carry out the same operation for all the simultaneously fitted spectra.
- Print - Press on the Print menu box with the left mouse button in order to carry out the default print operation assigned to the Print menu box in the corresponding submenu of the SET menu box.
Press on the Print menu box with the right mouse button in order to access one of the following options.
- - Select this option in order to print the textual fit report associated with one or more of the fitted spectra to the default printer (example). In order to send the print job to a different printer, select the printer in question from the appearing list.
- - Select this option in order to print the graph of the current spectrum to the printer set as default in the SET menu. In order to send the print job to a different printer, select the printer in question from the appearing list.
- - Select this option in order to print the graph of the current spectrum and those of the fitted distributions (if any) to the printer set as default in the SET menu. The graphs are all printed on the same page. In order to send the print job to a different printer, select the printer in question from the appearing list.
- - Select this option in order to print to the default printer the textual fit report(s), and then on separate page (pages) the graph (graphs) of the spectrum (spectra) and fitted distributions (if any). In order to send the print job to a different printer, select the printer in question from the appearing list.
- - Select this option in order to print to the default printer the graph of all the simultaneously fitted spectra, on a single page. In order to send the print job to a different printer, select the printer in question from the appearing list.
- - Select this option in order to copy the textual fit report associated with the current spectrum to the clipboard of Windows (example).
- - Select this option in order to copy the textual fit report of all the simultaneously fitted spectra to the clipboard of Windows. If a parameter (submenu item) is selected, then the individual fit reports will follow each other in order according to the value of the selected parameter.
- - Select this option in order to copy the fit results to the clipboard as comma-delimited text forming a table in which the parameters characterizing a particular spectrum are all listed in a single row, whereas each parameter - as well as each standard deviation value - has its own column (example).
This arrangement is especially useful for drawing the value of some parameter (e.g. isomer shift) as a function of another one (e.g. temperature) by the help of an external grapher program.
- - Select this option in order to copy the fit results to the clipboard as comma-delimited text forming a table in which the parameters characterizing a particular spectrum are all listed in a single column, whereas each parameter has its own row (example).
This arrangement is considered to be useful when creating tables for a presentation.
- - Select this option in order to copy the graph of the current spectrum to the clipboard of Windows according to the default settings attributed to Clipboard - single spectrum on the Printer Setup Dialog.
- - Select this option in order to copy the graph of the current spectrum and those of the fitted distributions (if any) to the clipboard of Windows.
- - Select this option in order to copy the graphical content of non-empty insight pages (if any) to the clipboard of Windows.
- - Select this option in order to copy the graph of all the simultaneously fitted spectra to the clipboard of Windows (example). If a parameter (submenu item) is selected, then the spectrum graphs will be arranged according to the value of the selected parameter.
- - Select this option in order to save and open a HTML FitLog for one or more of the fitted spectra according to the current state of the fit. The FitLog file is saved to the folder that contains also the corresponding spectrum data file, and it is opened in the default web browser application.
- - Select this option in order to save the fit report of the current spectrum as text file (example).
- - Select this option in order to save the fit report of all the simultaneously fitted spectra in the same text file one after the other.
- - Select this option in order to save the fit results as comma-delimited text into a text file forming a table in which the parameters characterizing a particular spectrum are all listed in a single row, whereas each parameter - as well as each standard deviation value - has its own column (example).
This arrangement is especially useful for drawing the value of some parameter (e.g. isomer shift) as a function of another one (e.g. temperature) by the help of an external grapher program.
- - Select this option in order to save the fit results as comma-delimited text into a text file forming a table in which the parameters characterizing a particular spectrum are all listed in a single column, whereas each parameter has its own row (example).
This arrangement is considered to be useful when creating tables for a presentation.
- Set - Press on the Set menu box in order to access one of the following options.
- - Turn to this option in order to set/modify spectrum parameters associated with the current spectrum.
- - Turn to this option in order to set/modify the isomer shift of the isomer shift reference material (associated with the current source nuclide) relative to the corresponding standard IS reference material.
The value given here is considered when the fitness of MIDB records is evaluated (with respect to the current spectrum) for example when the Find and apply best match submenu option of the DB menu is selected.
- - Turn to this option in order to constrain all line width type parameters of the current model to have always the same value,
or to make all line width type parameters independent. To impose more refined constraints of this type, turn to the Constrain menu.
- Constrain all to be same
- Set all to be independent
- - Turn to this menu to access the following options.
- Share for the selected parameter (The constraints imposed on the selected parameter will be set for the corresponding parameters in the simultaneously fitted spectra.)
- Share for position and width parameters (The constraints imposed on the position and width type parameters of the selected subspectrum will be set for the corresponding parameters in the simultaneously fitted spectra.)
- Share for all position and width parameters (The constraints imposed on the position and width type parameters of the current spectrum will be set for the corresponding parameters in the simultaneously fitted spectra.)
- Stretch to make current parameter values valid (For parameters whose value lies outside of the allowed range, the parameter boundaries will be extended such that the allowed range includes the current value of the parameter.)
- - Turn to this option in order to enable / disable mixing of the Lorentzian with dispersion line shape with other type of line shapes in the same model. Normally, in the case of interference with the atomic photoelectric effect (see PRL 28 (1972) 530 and
PRC 8 (1973) 1916), it is physically reasonable to fit all the absorption peaks with the Lorentzian with dispersion line shape.
- Set it only for the selected subspectrum
- Set it for all the subspectra
- - Normally, if transmission integral is used to fit a spectrum with more than one subcomponents, then subspectrum curves are formally indicated emphasizing the corresponding nuclear absorption spectrum. (In such a case, however, the fitting curve cannot be produced by the simple addition of the indicated subspectra.)
Select this option in order to enable / disable the calculation of such subspectra.
- - Turn to this option in order to determine the relative precision by which the fit parameters associated with a local optimum should be determined during the fit procedure.
- - Turn to these options in order to determine whether amplitude type parameters can take on negative values or not. Normally, amplitude type parameters should be forced to be non-negative.
- - Turn to these options in order to determine whether line width type parameters can take on negative values or not. Normally, line width type parameters should be forced to be positive. (Setting of negative line width values may lead to meaningless results.)
- - Select this option in order to erase the calibration of the velocity axis, i.e. to make the horizontal axis to display channel numbers instead of velocity values.
- - Select this option to set the internal resolution of the graphical user interface (GUI).
- - Select this option to set various configuration parameters influencing the operation of MossWinn.
- - Turn to this menu in order to determine what should happen when the Print box is pressed on by the left mouse button.
- Print fit results as text
- Print fit results and then the spectrum
- Print fit results then the spectrum + distributions
- Copy fit results to the clipboard
- Model - click on the Model tab in order to access the model page that allows the setting of the attributes of the fit model set for the current spectrum. The function of the various visual elements of the model page are listed below.

- Model groups popup box () - Press on this area with the left mouse button in order to access the model groups and the corresponding fit models that were saved earlier via the Save Model As menu. One can select here either a model group or directly one of the fit models. When multiple spectra are fitted simultaneously, the selected model can be set either for the current spectrum or for all the simultaneously fitted spectra at once. Originally only a single model group, named GENERAL, is defined. Each model group can contain around 32 fit models, and there can be around 32 model groups allowing to manage more than 1000 fit models altogether.
- Models popup box () - Press on this area with the left mouse button in order to access the models (saved earlier via the Save Model As menu) of the currently selected model group. When multiple spectra are fitted simultaneously, the selected model can be set either for the current spectrum or for all the simultaneously fitted spectra at once. Originally each model group contains only a single model, named CUSTOM. The CUSTOM model of a model group contains the fit model that was abandoned when the model group in question was selected.
- Source popup box () - Press on the source popup box to select the Mossbauer nuclide / transition that was used to measure the fitted spectra. The same nuclide / transition is set for all the simultaneously fitted spectra. If one of the transition types (, , ) is selected then the nuclear parameters (nuclear spin, g-factor, quadrupole moment, etc.) are added to the parameter list of the background as fixed parameters. When unfixed, the nuclear parameters can be fitted as well.
- Subspectrum popup box - Press on the subspectrum popup box in order to select one of the subspectra being part of the fit model set for the current spectrum. The parameters of the selected subspectrum will appear in the parameter list box. The parameter list of the spectrum background is formally treated here as the 0th subspectrum named .
- Parameter list box - This list box displays the parameters belonging to the subspectrum currently selected in the Subspectrum popup box. Press on the name of one of the parameters in order to make the corresponding parameter constraints become listed in the parameter constraints list box on the right. Press on the arrows or use the mouse wheel in order to scroll the list of parameters up or down. A parameter can also be selected by moving the mouse pointer over the corresponding navigation shape on the parameters panel.
- Parameter constraints list box - This list box displays the constraints imposed on the parameter currently selected in the parameter list box. Some of the constraints can be edited manually: press on such constraints with the left mouse button in order to edit them. In order to delete a constraint, erase all of its characters in edit mode. Press on a constraint with the right mouse button in order to disable / enable it, without deletion. A constraint is in effect (i.e. enabled) only if it is written with white color on a blue background. One cannot delete or disable all of the constraints: at least one of them has to remain in effect. Whereas parameter constraints are always obeyed by the Global Fit procedure, the local fit procedure respects only those constraints that are marked to be a Hard constraint by the help of the corresponding menu of the Constrain popup box. If for a parameter more than one constraint is defined, then the enabled constraints are evaluated from top to bottom one after the other. The result of the evaluation (i.e. the ranges enabled for the parameter) can be examined on the Details page. In the following examples of constraints are listed together with their meaning.
- - the parameter has to remain in the range of 0.1 – 1.2 .
- - the parameter has to be equal to -3.4.
- - the parameter can take on values in the range of 45 – 55 .
- - the parameter cannot take on values of the range 50 – 52 .
- - the parameter should be higher than 1.5 .
- - the parameter should be less or equal 2.1 .
- - the parameter constraints should be obeyed also by the local fit procedure.
- If all the four of the above constraints are defined in this order for a single parameter, then the parameter in question can take on values only in the range of [0,0.1] ∪ [0.2,0.5) ∪ [0.6,0.7] ∪ (1,1.2].
- Interaction popup box - Press on this popup box in order to select the — built-in or user-written external (DLL) — theoretical model to be applied to the current subspectrum. Built in theoretical models available, e.g., for 57Fe are listed below.
- - a single absorption peak (singlet).
- - quadrupole doublet consisting of two peaks of equal amplitudes and line widths. Linear model, it can be altered on the Details page.
- - magnetic sextet consisting of six peaks having equal line widths and relative amplitudes of 3:2:1:1:2:3. Linear model, it can be altered on the Details page.
- - magnetic sextet consisting of six peaks having equal line widths and relative amplitudes of 3:2:1:1:2:3. The sextet takes quadrupole interaction into account by treating the quadrupole interaction as a small perturbation of the magnetic one:
the two outermost lines of the sextet are shifted along the velocity axis by +Δ/2 whereas the four inner lines are shifted by -Δ/2, where Δ is the quadrupole splitting. Linear model, it can be altered on the Details page.
- - in general 8 peaks with equal line width, having positions and amplitudes calculated by solving the static Hamiltonian of mixed magnetic and quadrupole interactions. The ez basis vector of the reference coordinate system (figure) is assumed to be parallel to the hyperfine magnetic field.
- - in general 8 peaks with equal line width, having positions and amplitudes calculated by solving the static Hamiltonian of mixed magnetic and quadrupole interactions (as described in NIM 48 (1967) 219). The ez basis vector of the reference coordinate system (figure) is assumed to be parallel to the z axis of the eigensystem of the EFG.
- - model of uniaxial superparamagnetic relaxation as described in PR 165 (1968) 446.
- - model considering quadrupole interaction in the presence of a fluctuating electric field gradient, expected e.g. in the case of the dynamic Jahn-Teller effect. Described in PR 165 (1968) 456.
- - model implemented for 57Fe in paramagnetic powder where electron hopping occurs either between iron ions of different oxidation states (e.g. Fe2+ ↔ Fe3+) or between iron ions and the rest of the system. It can model situations encountered, e.g., in Phys. Chem. Minerals 10 (1984) 250. and PRB 31 (1985) 34.
- - model implemented for 57Fe in a magnetic powder where electron hopping occurs either between iron ions of different oxidation states (e.g. Fe2+ ↔ Fe3+) or between iron ions and the rest of the system. It can model situations encountered, e.g., in J. Phys. Chem. Solids 41 (1980) 1273.
- - model of a powdered sample with randomly oriented EFG (displaying zero asymmetry parameter) and uniaxial hyperfine magnetic field, as described in NIM B 9 (1985) 201.
- - paramagnetic hyperfine structure models, as listed below. These models require either the DBM version of MossWinn or subscription to the MossWinn services. See examples and the manual for further details.
- - accounts for a powder sample with an electronic system with effective spin S = ½, by assuming slow electronic relaxation in the absence of an external magnetic field. The eigensystem of the hyperfine magnetic interaction tensor (A) and the electric field gradient tensor (EFG) are assumed to be coincident. See, for example, Phys. Rev.152 (1966) 345.
- - accounts for a powder sample with an electronic system with effective spin S = 0...5/2, by assuming slow electronic relaxation in the absence of an external magnetic field. The eigensystem of the hyperfine magnetic interaction tensor (A) and the electric field gradient tensor (EFG) are assumed to be coincident with the x,y,z crystal field system (CFS). See, for example, Rep. Prog. Phys. 16 (1953) 108-159. and Phys. Rev. 148 (1966) 211.
- - accounts for a powder sample with an electronic system with effective spin S = 0...5/2, by assuming slow electronic relaxation in the absence of an external magnetic field. The eigensystem of the hyperfine magnetic interaction tensor (A) and the electric field gradient tensor (EFG) are assumed to be coincident with the x,y,z crystal field system (CFS) whose z axis is oriented in the (1,1,1) cube diagonal direction of the cubic crystal field system (trigonal distortion). Tensor A is assumed to display axial symmetry (Ayy = Axx). See, for example, Biochemistry (Structure and Bonding) 20 (1974) 59..
- - accounts for a powder sample with an electronic system with effective spin S = 0...5/2, by assuming slow electronic relaxation in the absence of an external magnetic field. The eigensystem of the hyperfine magnetic interaction tensor (A) and the electric field gradient tensor (EFG) are given with respect to the x,y,z crystal field system (CFS) via the respective Euler angles that rotate the CFS system into the A or EFG tensor eigensystem. The orientation of the x,y,z CFS system is given with respect to the [100, 010, 001] cubic system via the Euler angles that rotate the cubic system into the CFS system.
- - accounts for a powder sample with an electronic system with effective spin S = ½, by assuming slow electronic relaxation in an external magnetic field oriented either parallel or perpendicular to the Mössbauer γ-ray direction. The eigensystem of the electron gyromagnetic tensor (g), the hyperfine magnetic interaction tensor (A) and the electric field gradient tensor (EFG) are assumed to be coincident.
- - accounts for a powder sample with an electronic system with effective spin S = 0...5/2, by assuming slow electronic relaxation in an external magnetic field oriented either parallel or perpendicular to the Mössbauer γ-ray direction. The eigensystem of the electron gyromagnetic tensor (g), the hyperfine magnetic interaction tensor (A) and the electric field gradient tensor (EFG) are assumed to be coincident with the x,y,z crystal field system (CFS).
- - accounts for a powder sample with an electronic system with effective spin S = 0...5/2, by assuming slow electronic relaxation in an external magnetic field oriented either parallel or perpendicular to the Mössbauer γ-ray direction. The eigensystem of the electron gyromagnetic tensor (g), the hyperfine magnetic interaction tensor (A) and the electric field gradient tensor (EFG) are assumed to be coincident with the x,y,z crystal field system (CFS) whose z axis is oriented in the (1,1,1) cube diagonal direction of the cubic crystal field system (trigonal distortion). Tensors g and A are assumed to display axial symmetry (gyy = gxx, Ayy = Axx).
- - accounts for a powder sample with an electronic system with effective spin S = 0...5/2, by assuming slow electronic relaxation in an external magnetic field oriented either parallel or perpendicular to the Mössbauer γ-ray direction. The eigensystem of the electron gyromagnetic tensor (g), the hyperfine magnetic interaction tensor (A) and the electric field gradient tensor (EFG) are given with respect to the x,y,z crystal field system (CFS) via the respective Euler angles that rotate the CFS system into the g, A or EFG tensor eigensystem. The orientation of the x,y,z CFS system is given with respect to the [100, 010, 001] cubic system via the Euler angles that rotate the cubic system into the CFS system.
- - accounts for a powder sample with an electronic system with effective spin S = ½ being decoupled from the nuclear spin states, by assuming fast electronic relaxation in an external magnetic field oriented either parallel or perpendicular to the Mössbauer γ-ray direction. The eigensystem of the electron gyromagnetic tensor (g), the hyperfine magnetic interaction tensor (A) and the electric field gradient tensor (EFG) are assumed to be coincident.
- - accounts for a powder sample with an electronic system with effective spin S = 0...5/2 being decoupled from the nuclear spin states, by assuming fast electronic relaxation in an external magnetic field oriented either parallel or perpendicular to the Mössbauer γ-ray direction. The eigensystem of the electron gyromagnetic tensor (g), the hyperfine magnetic interaction tensor (A) and the electric field gradient tensor (EFG) are assumed to be coincident with the x,y,z crystal field system (CFS).
- - accounts for a powder sample with an electronic system with effective spin S = 0...5/2 being decoupled from the nuclear spin states, by assuming fast electronic relaxation in an external magnetic field oriented either parallel or perpendicular to the Mössbauer γ-ray direction. The eigensystem of the electron gyromagnetic tensor (g), the hyperfine magnetic interaction tensor (A) and the electric field gradient tensor (EFG) are assumed to be coincident with the x,y,z crystal field system (CFS) whose z axis is oriented in the (1,1,1) cube diagonal direction of the cubic crystal field system (trigonal distortion). Tensors g and A are assumed to display axial symmetry (gyy = gxx, Ayy = Axx).
- - accounts for a powder sample with an electronic system with effective spin S = 0...5/2 being decoupled from the nuclear spin states, by assuming fast electronic relaxation in an external magnetic field oriented either parallel or perpendicular to the Mössbauer γ-ray direction. The eigensystem of the electron gyromagnetic tensor (g), the hyperfine magnetic interaction tensor (A) and the electric field gradient tensor (EFG) are given with respect to the x,y,z crystal field system (CFS) via the respective Euler angles that rotate the CFS system into the g, A or EFG tensor eigensystem. The orientation of the x,y,z CFS system is given with respect to the [100, 010, 001] cubic system via the Euler angles that rotate the cubic system into the CFS system.
- - distribution models with parameterized profile, as listed below. See the manual for further details.
- - doublet spectrum model of a paramagnetic powder with a Gaussian distribution in the quadrupole splitting, and with an associated Gaussian distribution in the isomer shift values, the latter being coupled to the quadrupole splitting values according to a linear relationship. It is based on Voigt-based fitting (VBF) as described in NIM B 58 (1991) 85.
- - sextet spectrum model of a magnetic powder with a Gaussian distribution in the hyperfine magnetic field, and with an associated Gaussian distribution in the isomer shift and quadrupole splitting values, the latter ones being coupled to the hyperfine magnetic field values according to linear relationships. It is based on Voigt-based fitting (VBF) as described in NIM B 58 (1991) 85. In this model the quadrupole interaction is taken into account in the first-order perturbation limit.
- - doublet spectrum model of a paramagnetic powder with a Gaussian distribution in the quadrupole splitting and isomer shift values, with an arbitrary degree of correlation between the two distributions. It is based on extended Voigt-based fitting (xVBF) as described in NIM B 129 (1997) 266.
- - sextet spectrum model of a magnetic powder with a Gaussian distribution in the isomer shift, hyperfine magnetic field and quadrupole splitting values, with an adjustable degree of pairwise correlations between the different distributions. It is based on extended Voigt-based fitting (xVBF) as described in NIM B 129 (1997) 266. In this model the quadrupole interaction is taken into account in the first-order perturbation limit.
- - sextet-based spectrum model of a binomial atomic distribution in a magnetic powder sample in which the isomer shift, hyperfine magnetic field and quadrupole splitting parameters are perturbed in an additive manner by substituent atoms distributed randomly in the first shell of atomic neighbors around the Mössbauer nuclides. Each substituent atom in the shell is assumed to alter the isomer shift, hyperfine magnetic field and quadrupole splitting parameters with the same amount. In this model the quadrupole interaction is taken into account in the first-order perturbation limit. The model is also suitable to fit doublet- or singlet-based binomial atomic distribution models.
- - sextet-based spectrum model of a binomial atomic distribution in a magnetic powder sample in which the isomer shift, hyperfine magnetic field and quadrupole splitting parameters are perturbed in an additive manner by substituent atoms distributed randomly in the first and second shells of atomic neighbors around the Mössbauer nuclides. Substituent atoms in the same shell are assumed to alter the isomer shift, hyperfine magnetic field and quadrupole splitting parameters with the same amount, but this amount can be different for atoms in different shells. In this model the quadrupole interaction is taken into account in the first-order perturbation limit. The model is also suitable to fit doublet- or singlet-based binomial atomic distribution models.
- - sextet-based spectrum model of a multinomial atomic distribution in a magnetic powder sample in which the isomer shift, hyperfine magnetic field and quadrupole splitting parameters are perturbed in an additive manner by two different substituent atoms of the type 'A' and 'B' distributed randomly in the first shell of atomic neighbors around the Mössbauer nuclides. Substituent atoms of the same type are assumed to alter the isomer shift, hyperfine magnetic field and quadrupole splitting parameters with the same amount, but the amount in question may be different for 'A' and 'B' type substituent atoms. In this model the quadrupole interaction is taken into account in the first-order perturbation limit. The model is also suitable to fit doublet- or singlet-based multinomial atomic distribution models.
- - quadrupole doublet consisting of two peaks of equal line widths and independent amplitudes. Linear model, it can be altered on the Details page.
- - magnetic sextet consisting of six peaks having equal line widths and relative amplitudes of 3:x:1:1:x:3 where x is an arbitrary non-negative value. Linear model, it can be altered on the Details page.
- - magnetic sextet consisting of six peaks having equal line widths and relative amplitudes of 3:x:1:1:x:3 where x is an arbitrary non-negative value. The sextet takes quadrupole interaction into account by treating the quadrupole interaction as a small perturbation of the magnetic one:
the two outermost lines of the sextet are shifted along the velocity axis by +Δ/2 whereas the four inner lines are shifted by -Δ/2, where Δ is the quadrupole splitting. Linear model, it can be altered on the Details page.
- - in general 8 peaks with equal line width, having positions and amplitudes calculated by solving the static Hamiltonian of mixed magnetic and quadrupole interactions. The ez basis vector of the reference coordinate system (figure) is assumed to be parallel to the hyperfine magnetic field. A single crystal is assumed, i.e. the relative amplitudes of the absorption peaks are influenced by the orientation of the γ-ray direction.
- - in general 8 peaks with equal line width, having positions and amplitudes calculated by solving the static Hamiltonian of mixed magnetic and quadrupole interactions (as described in NIM 48 (1967) 219). The ez basis vector of the reference coordinate system (figure) is assumed to be parallel to the z axis of the eigensystem of the EFG. A single crystal is assumed, i.e. the relative amplitudes of the absorption peaks are influenced by the orientation of the γ-ray direction.
- - in general 8 peaks with equal line width, having positions and amplitudes calculated by solving the static Hamiltonian of mixed magnetic and quadrupole interactions. The ez basis vector of the reference coordinate system (figure) is assumed to be parallel to the hyperfine magnetic field. Mosaic sample is assumed, i.e. the azimuthal angle of the gamma-ray direction is assumed to be random and evenly distributed between 0 deg and 360 deg around the ez basis vector.
- - in general 8 peaks with equal line width, having positions and amplitudes calculated by solving the static Hamiltonian of mixed magnetic and quadrupole interactions. The ez basis vector of the reference coordinate system (figure) is assumed to be parallel to the z axis of the eigensystem of the EFG. Mosaic sample is assumed, i.e. the azimuthal angle of the gamma-ray direction is assumed to be random and evenly distributed between 0 deg and 360 deg around the ez basis vector.
- - in general 8 peaks with equal line width, having positions and amplitudes calculated by solving the static Hamiltonian of mixed magnetic and quadrupole interactions in the presence of the Goldanskii-Karyagin effect. The ez basis vector of the reference coordinate system (figure) is assumed to be parallel to the hyperfine magnetic field. The MSD (mean square displacement) tensor is assumed to be diagonal in the reference coordinate system, and furthermore it is assumed to display axial symmetry.
- - in general 8 peaks with equal line width, having positions and amplitudes calculated by solving the static Hamiltonian of mixed magnetic and quadrupole interactions (as described in NIM 48 (1967) 219) in the presence of the Goldanskii-Karyagin effect. The ez basis vector of the reference coordinate system (figure) is assumed to be parallel to the z axis of the eigensystem of the EFG. The MSD (mean square displacement) tensor is assumed to be diagonal in the reference coordinate system, and furthermore it is assumed to display axial symmetry. See NIM 136 (1976) 569 and Appl. Phys. 1 (1973) 93 for details.
- Peak shape popup box - Press on this popup box in order to select the peak shape to be applied to the currently selected subspectrum. All the peaks belonging to the subspectrum will have the same peak shape. The following peak shapes are available.
- - Lorentzian peak shape described by three parameters, A (denoting either the amplitude or the area of the peak), IS (denoting the center of the peak), and Γ (denoting the FWHM of the peak).
Depending om whether Amplitude or Area mode is selected, the peak is calculated as
- - Approximation of the Voigt line profile, calculated as described in J. APPL. CRYST. 19 (1986) 63.
When the Pseudo-Voigt line shape is selected for a subspectrum, then the former line width parameter(s) will determine the line width (FWHM) of the associated Gaussian profile(s), whereas the Lorentzian line width (being common to all the peaks belonging to the same subspectrum) is added as an additional parameter to the parameter list of the subspectrum in question.
- - Peak shape that takes into account the interference effect between nuclear and atomic absorption of γ-rays (see PRL 28 (1972) 530 and
PRC 8 (1973) 1916 for details), described by four parameters, A (denoting either the amplitude or the area of the peak), IS (denoting the center of the peak), Γ (denoting the FWHM of the peak) and ξ (denoting the dispersion amplitude):
When Lorentzian with dispersion is selected as peak shape for one of the subspectra, then the ξ dispersion amplitude (being common to all the subspectra belonging to the same spectrum) is added as an additional parameter to the parameter list of the background.
- - Peak shape that takes into account the cosine-broadening effect caused by non-ideal geometrical conditions. The peak shape is calculated as described in
Hyp. Int. 29 (1986) 1539. When this line shape is selected for a subspectrum, a new parameter named Visual angle [deg], denoting the visual angle of the absorber or the detector aperture (which is smaller depending on the actual geometrical conditions) viewed from the center of the source, is added to the parameter list of the subspectrum in question.
In the case of a Lorentzian with cosine smearing, the amplitude, area and percentage values displayed or printed by MossWinn are exactly those which one would get for the corresponding Lorentzian without cosine smearing.
That is (apart from possible f-factor differences) subspectrum relative areas in this case also reflect the relative occurrence of Mossbauer nuclides in different microenvironments. At the same time the area values are not equal to the corresponding apparent (measured and fitted) spectral area.
- Thin absorber limit () / Transmission Integral () / TI source function... () - Press on this popup box in order to choose between thin absorber approximation and transmission integral modes. When transmission integral mode is selected, the necessary additional parameters (, , , ) are automatically added to the parameter list of the background. In transmission integral mode, the usual subspectrum line widths denote absorber line widths.
In the different transmission integral modes, the source line shape is expressed as follows (Γs denotes the source line width):
Transmission Integral Mode | Source line shape |
| |
| |
|
- Add - The Add menu box provides access to functions related to the following kinds of addition operations.
- - This menu provides access to subspectrum models saved earlier via the Save Subspectrum menu option. Select one of the subspectrum models in order to add a corresponding subspectrum to the current fit model.
Subspectra are listed by their entry names that may be different from the actual name given to the subspectrum when it is added to the current model. If a subspectrum with the same name already exists, then the name of the new subspectrum will be made unique by numbering.
Press on the menu item in order to add a singlet to the current model. Note that subspectra that are already part of the fit model can be duplicated by turning to the option of their headline menu.
- - This menu provides access to subspectrum models available in — user programmed and compiled — dynamic link libraries being saved in the code library directory with a name corresponding to the mask .
(DLL libraries are listed here only if they were enabled in the SET menu.) Select one of the subspectrum models in order to add a corresponding subspectrum to the current fit model.
- - This menu provides access to subspectrum group models saved earlier via the Save Subspectrum Group... menu option. Select one of the subspectrum group models in order to add a corresponding subspectrum group to the current fit model.
Subspectrum groups are a collection of several subspectra that may be connected to eachother by user-defined constraints. A subspectrum group can also be used to model a phase whose Mossbauer spectrum consists of more than one subspectrum. For example, at room temperature the phase magnetite may be modeled by two sextets having an area ratio of 2:1 corresponding to iron atoms at octahedral and tetrahedral sites, respectively. Thus, room-temperature magnetite may be described by a subspectrum group consisting of two sextets with a fixed area ratio of 2:1.
- - Turn to this option in order to add the selected subspectrum (as a shared subspectrum) to all the fit models fitted simultaneously.
- - Turn to this option in order to add the selected DLL subspectrum (as a shared subspectrum) to all the fit models fitted simultaneously.
- - Turn to this option in order to add the selected subspectrum group (as a group of shared subspectra) to all the fit models fitted simultaneously.
- - Turn to this option in order to add an extra (custom) fit parameter to the parameter list of the background. Extra parameters may be used to describe experimental variables of the measurement, e.g. temperature, external magnetic field, sample orientation etc. Fit models may be made to depend on the extra parameters such that theoretical dependencies may be taken into account. For example, isomer shift parameters may be enforced to decrease linearly with temperature thereby modeling (approximately) the effect of the second order doppler shift at higher temperatures.
(Dependency functions of arbitrary complexity can be defined by modifying the DEP_DLL1.PAS source and compiling it to observe a new DEP_DLL1.DLL dynamic link library.)
The value of fit parameters can also be examined/tabulated as a function of extra parameters: for example, the value of a hyperfine magnetic field parameter may be depicted as a function of temperature or external magnetic field on one of the insight pages.
- - Turn to this option in order to add one or more new spectra (selected among those being loaded on the current desk in the main menu) to the fit. Spectra added to the fit are all fitted simultaneously. For the newly added spectra the fit model set for the current spectrum will be initiated. To remove a spectrum from the list of simultaneously fitted spectra, turn to the Remove menu box.
- Remove - The Remove menu box provides access to functions related to the following kinds of removal operations.
- - Turn to this option in order to remove one of the shared or unshared subspectra from the fit model. A shared subspectrum can be removed from the current model as well as from models set for other simultaneously fitted spectra.
- - Turn to this option in order to remove all shared and unshared subspectra from the fit model associated with the current spectrum. Select the appearing submenu item in order to carry out the same operation for all the simultaneously fitted spectra.
- - Turn to this option in order to remove one of the spectra from a simultaneous fit.
- - Turn to this option in order to delete one of the subspectra saved earlier via the Save Subspectrum menu option to the subspectrum collection.
- - Turn to this option in order to delete one of the subspectrum groups saved earlier via the Save Subspectrum Group... menu option.
- - Turn to this option in order to delete one of the fit models (of the current model group) saved earlier via the Save Model As menu option.
- - Turn to this option in order to delete one of the model groups with all of its models.
- - Turn to this option in order to delete the HTML FitLog file created earlier for the current spectrum.
- - Turn to this option in order to delete one of the parameters of the currently selected subspectrum.
- Save - The Save menu box provides access to functions related to the following kinds of save operations.
- - Turn to this option in order to save one of the subspectra to the subspectrum collection. The name of the subspectrum (as displayed by the subspectrum headline) as well as its entry name (by which it is identified in the subspectrum collection) can be defined after the subspectrum is selected. Turn to the Add menu box in order to add the saved subspectrum to a fit model.
- - Turn to this option in order to save two or more subspectra of the current model as a subspectrum group. The individual subspectra will be saved with their current name. The constraints (if any) among the parameters of the saved subspectra will be preserved in the saved subspectrum group. Turn to the Add menu box in order to add the saved subspectrum group to a fit model.
- - Turn to this option in order to save the current model to an existing or to a newly created model group. The saved model can be reloaded either via the Models popup box or the Model groups popup box.
- - Turn to this option in order to save an additional HTML FitLog to the FitLog file of the current spectrum, according to the current state of the fit. The FitLog file is located/created in the folder that contains also the corresponding spectrum data file. Its name is derived from the file name of the latter via the addition of . Multiple FitLogs can be saved to a single FitLog file. MossWinn takes care of duplicates: it will not add a new FitLog if it is essentially identical with one being already present in the FitLog file. The FitLog file can be opened by pressing on the gray info line popup box directly below the spectrum graph area. To access the functions related to the FitLog file, press with the right mouse button on the Accept menu box.
- - Turn to this option in order to save an independent copy of the current spectrum with an arbitrary path and name. (The fit will continue with the original spectrum.)
- - Turn to this option in order to save the content of one of the active insight pages as a textual data file.
- Details - Press on the Details menu box in order to invoke the Details panel that provides access - among others - to the matrix elements influencing the amplitudes, positions and widths of the peaks contributing to a linear model. For details press on one of the visual elements on the image below.

- - Press on this popup box to select the subspectrum to handle.
- - This box lists the parameters associated with the selected subspectrum. Press on any of the parameter names in order to select it, and to have the lines affected by the parameter in question become selected in the list on the right.
- - This box lists the Lines (peaks) associated with the selected subspectrum. The Lines affected by the selected parameter are displayed blue-selected. In case of Amplitude and Line Width type parameters press on any of the Lines to have it selected/deselected. The number and name of the corresponding Amplitude and Line Width type parameters will be adjusted automatically as needed.
- - This box lists the matrix elements associated with the selected parameter. The matrix elements reflect how the selected amplitude/position/width type parameter affects the amplitude/position/width of the absorption peaks associated with the subspectrum. For example the case shown above reflects that the value of the quadrupole splitting parameter is multiplied by / before the result is added to the position of / . Press on any of the matrix elements in order to edit the corresponding value.
- - Press on the Hide box in order to hide the details panel.
- - Press on the Tools popup box in order to alter the linear model - associated with the selected subspectrum - via the following options.
- - Turn to this option in order to add a position type parameter to the linear model.
- - Turn to this option in order to remove one of the position type parameters of the linear model.
- - Turn to this option in order to add an absorption line to the linear model.
- - Turn to this option in order to remove one of the absorption lines of the linear model.
- - Press on this popup box in order to access the following functions of the details panel.
- - Select this option in order to have the details panel display the affected lines and the matrix elements associated with the selected parameter, as in the example shown above.
- - Select this option in order to have the details panel display the enabled intervals associated with the selected parameter. The enabled intervals are calculated on the basis of the constraints displayed in the Parameter constraints list box.
- - Select this option in order to have the details panel display the current value of internal program variables associated with the selected subspectrum and parameter. The details of this feature are not documented: during development/testing it is used by the author to monitor the internal state of some relevant program variables.
- - Turn to this option in order to search through the database records in order to find the spectrum that best matches the fitted spectrum currently on the screen, and to set the corresponding fit model as included in the database record returned by the search. (The currently set fit model will be overwritten.)
The fitness of the database records with respect to the current spectrum is evaluated by considering the isomer shift of the isomer shift reference material (associated with the current source nuclide) that can be set via the Isomer shift reference submenu option of the SET menu.
The search can be performed via the following submenu options (a press directly on the menu item is equivalent to the option ):
- - The search will consider only those records that were published in the database by using the hardware key that is currently attached to the computer.
- - The search will consider all database records.
- - The search will consider only those database records that refer to measurements carried out in transmission (reflection) geometry.
- The fit model returned as the best match will be saved automatically as a new model of the model group where stands for the corresponding source nuclide. Apart from the best match, the search will also return several further records in descending order of the fitness of their included fit model with respect to the spectrum under analysis. Select this menu again in order to see the corresponding list and apply another one of the returned records.
- - Turn to this option in order to select one of the records returned by the Find and apply best match function, and open the corresponding compound class in the database browser.
- - Select this option in order to display the database browser that allows one to search through the database records according to various criteria. The database browser also provides the possibility to try/apply the fit models, represented by the database records, to the fitted spectrum currently on the screen.
- - Select this option in order to publish the current fit model and/or a downsampled version of the current spectrum in the MossWinn Internet Database. If along with the fit parameters you also want to publish their standard deviation in the database, calculate the corresponding standard deviations via the Cal StD menu before turning to this option.
Once the option is selected, first the Database record input form becomes displayed on which one can set various parameters associated with the record to be published. Then, the next form provides the possibility to set the resampled form of the measured spectrum, and to declare whether one wants to publish only the fit model, only the resampled spectrum, or both. On the next form a preview of the record is displayed, where one can check how the record will appear when published. The record becomes finally published when on the preview form the Publish record button is pressed.
Once a record is published in the database, it may appear in the MIDB database browser of MIDB subscribers all around the world. The MIDB Publisher's Guide contains useful information on recommended practices concerning the publication of MIDB records.
- - Select this option in order to withdraw a MIDB record published earlier by using the hardware key that is currently attached to the computer ( own record ). The own record that is to be withdrawn can be selected on the appearing form. Withdrawal of own records from the MIDB database does not require subscription to the database.
- - Select this option to edit one of the own MIDB records by changing the record parameters that can be set on the Database record input form. The fit model and the resampled spectrum associated with the record cannot be edited in this way. The own record that is to be edited can be selected on the appearing form.
- - Select this option to carry out pending MIDB publication and withdrawal operations, and to synchronize local data with the MIDB host server. The frequency of such synchronization events may be subject to a limitation.
- - Select this option to open the MIDB Compound Summary in the default browser.
- - Select this option to bring up this html help concerning the DB menu box.
- Constrain - Press on the Constrain popup box in order to declare a new constraint for the selected fit parameter. Some of the constraints take effect immediately, others are effective only during fitting. The local fit procedure obeys these latter constraints only if they are designated to be constraints. Also, constraints take effect immediately.
Some of the constraints are initialized without being made active. Inactive constraints are listed without blue highlight in the Parameter constraints list box. Press on them with the right mouse button in order to make them active.
- (available only for position type parameters) - Declare a position type parameter to be of the distribution type in order to fit a distribution in the selected parameter (distribution parameter). For example hyperfine magnetic field distribution, quadrupole splitting distribution or isomer shift distribution can be fitted in this way. In order to declare correlations, constrain another parameter(s) to depend linearly on the distribution parameter.
The distribution curve can be made to be displayed (example) on either of the insight pages.
The following distribution types are available:
- Unrestricted (UNR) - The distribution is calculated as described in J. Phys. E: Sci. Instrum. 7 (1974) 526. The distribution curve can take on negative values as well.
- Positive, Free Boundaries (PFB) - Similar to the previous case, but the distribution cannot take on negative values.
- Positive, Zero Left Boundary (PZLB) - The distribution cannot take on negative values, and it goes to zero at its left boundary.
- Positive, Zero Right Boundary (PZRB) - The distribution cannot take on negative values, and it goes to zero at its right boundary.
- Positive, Zero Left & Right Boundaries (PZLRB) - The distribution cannot take on negative values, and it goes to zero at its both boundaries.
- Split distribution - Select this option in order to split the distribution at some intermediate value by adding the directive to the parameter constraints list box, where x denotes an intermediate distribution-parameter value. As a result, the distribution will contribute to the fitting curve by two separate subspectra: one belonging to distribution-parameter values higher than x, and the other belonging to distribution-parameter values lower than or equal to x.
-
When distributions are fitted in conjunction with transmission integral fitting then the parameter is automatically added to the parameter list of the background.
This parameter controls the level of noise filtering necessary for the calculations. Its value is constrained to the interval of [0,1]. A value of 1.0 means hardly any filtering, whereas a value of 0.0 means strongly suppressed noise level.
The parameter should be fitted in order to achieve optimal results. See the manual and corresponding publications for further details.
- (available only for amplitude type parameters) - Constrain an amplitude type parameter to be relative in order to fit its relative value compared to another parameter. If the amplitude type parameter - that is constrained to be relative - is the first amplitude type parameter in its subspectrum, then its absolute value is calculated as the product of its relative value and the parameter of the background.
Otherwise, the absolute value is calculated as the product of the relative value and the absolute value of the subspectrum's first amplitude type parameter.
For example, declaring the amplitude of the 2nd and 5th lines of a 57Fe sextet to be relative enables one to set the amplitude of the 2nd and 5th lines relative to that of the 3rd and 4th. In order to remove the Relative constraint, remove the word from the corresponding list of constraints.
- - The parameter should remain equal to the value that is to be given in the Parameter constraints list box.
- - The parameter should remain inside the range that is to be given in the Parameter constraints list box.
- - The parameter should be allowed to take on values from the range that is to be given in the Parameter constraints list box. This constraint can be used to modify the effect of other constraints.
- - The parameter should not be allowed to take on values from the range that is to be given in the Parameter constraints list box. This constraint can be used to modify the effect of other constraints.
- - The parameter should exceed the value that is to be given in the Parameter constraints list box. This constraint can be used to modify the effect of other constraints.
- - The parameter should not be less than the value that is to be given in the Parameter constraints list box. This constraint can be used to modify the effect of other constraints.
- - The parameter should be less than the value that is to be given in the Parameter constraints list box. This constraint can be used to modify the effect of other constraints.
- - The parameter should not exceed the value that is to be given in the Parameter constraints list box. This constraint can be used to modify the effect of other constraints.
- - The above type of constraints declared for the selected parameter should be obeyed also by the local fit procedure. Otherwise these constraints are taken into account only by the Global fitting procedure.
- - The parameter should exceed the one selected in the appearing submenu list.
- - The parameter should not be less than the one selected in the appearing submenu list.
- - The parameter should remain below the one selected in the appearing submenu list.
- - The parameter should not exceed the one selected in the appearing submenu list.
- - The parameter should not be equal to the one selected in the appearing submenu list.
- - The parameter should remain close to the one selected in the appearing submenu list. What is close and what is far is determined by the allowed parameter range of the latter parameter.
- - The parameter should remain far from the one selected in the appearing submenu list. What is close and what is far is determined by the allowed parameter range of the latter parameter.
- - The parameter should be equal to the one selected in the appearing submenu list. In order to remove the constraint, in the parameters' panel press on the name of the constrained parameter (displayed by brown color).
- - The parameter should be equal to the one selected in the appearing submenu list plus another, newly created parameter, which latter will be added to the parameter list of the subspectrum of the constrained parameter with the name where NAME is the name of the constrained parameter. In order to remove the constraint, in the parameters' panel press on the name of the constrained parameter (displayed by brown color).
- - The parameter should be equal to the product of the one selected in the appearing submenu list and another, newly created parameter, which latter will be added to the parameter list of the subspectrum of the constrained parameter with the name where NAME is the name of the constrained parameter. In order to remove the constraint, in the parameters' panel press on the name of the constrained parameter (displayed by brown color).
- - The parameter should depend linearly on the one selected in the appearing submenu list. The linear coefficient and the constant term will be added to the parameter list of the subspectrum of the constrained parameter as newly created parameters with the name and , respectively, where NAME is the name of the constrained parameter. In order to remove the constraint, in the parameters' panel press on the name of the constrained parameter (displayed by brown color).
- - The parameter should depend via the following — smooth and monotonic — function on the parameter (x) selected in the appearing submenu list:
Smooth monotonic function | Examples |
| |
|
The , , and parameters will be added to the parameter list of the subspectrum of the constrained parameter as newly created parameters with the names , , and , respectively, where NAME is the name of the constrained parameter. In order to remove the constraint, in the parameters' panel press on the name of the constrained parameter (displayed by brown color). This option can be used to set up a versatile constraint that can ensure the smooth, monotonic (increasing or decreasing) dependence of the currently selected fit parameter (y) on a physical parameter (x) such as the temperature of the sample, but may also model a step-like transition of a parameter as the function of another one.
The option requires either the DBM version of MossWinn or subscription to the MossWinn services.
- - The parameter should depend via the following — smooth, but not necessarily monotonic — function on the parameter (x) selected in the appearing submenu list:
Smooth function with slope | Examples |
| |
|
The , , , and parameters will be added to the parameter list of the subspectrum of the constrained parameter as newly created parameters with the names , , , and , respectively, where NAME is the name of the constrained parameter. In order to remove the constraint, in the parameters' panel press on the name of the constrained parameter (displayed by brown color). This option can be used to set up a versatile constraint that can ensure the smooth dependence of the currently selected fit parameter (y) on a physical parameter (x) such as the temperature of the sample, but may also model a step-like transition of a parameter as the function of another one.
The option requires either the DBM version of MossWinn or subscription to the MossWinn services.
- - The parameter should depend on the one selected in the appearing submenu list as determined by the - arbitrary - function included in the DEP_DLL1.DLL dynamic link library. (By default this is a cubic function.) The necessary number of additional parameters will be added to the parameter list of the subspectrum of the constrained parameter as newly created parameters with names determined by the functions in the DLL. In order to remove the constraint, in the parameters' panel press on the name of the constrained parameter (displayed by brown color).
- - Select this option in order to constrain the spectral area of the selected subspectrum by a percentage parameter that determines the relative spectral area of the subspectrum. The newly created percentage parameter will be added to the parameter list of the subspectrum whose area is constrained. In order to remove the percentage constraint, in the parameters' panel press on the name of the first amplitude type parameter (displayed by brown color) of the constrained subspectrum.
- - to copy all simultaneously fitted spectra to the clipboard as a single image (example). The spectra will follow each other according to the spectrum order set previously via the spectrum number popup box.
The resolution of the image can be set via the Printer Setup Dialog with selected as printer.
- - to copy all simultaneously fitted spectra to the clipboard as text, with the velocity values, the measured data, the fit envelope and the subspectra included as comma delimited columns. The spectra will follow each other according to the spectrum order set previously via the number of spectra popup box.
This option may be used to export numerical data from MossWinn for use in external data processing & graphics software.
- - to make a copy of all the simultaneously fitted spectra, all the valid insight pages, the fit results in two different tabular forms as well as the fit reports associated with the current state of the fit in the form of a single XLS spreadsheet file, in which the different spectra/insight pages/tables and the fit reports appear on separate worksheet pages (example).
This option provides a convenient way to collect the numerical data of the current state of the fit in a single XLS file for backup, graph creation or publication purposes. In order to have the standard deviation of fit parameters included in the XLS file, turn to the Cal StD function before selecting this option. Once created, MossWinn attempts to open the XLS file with an associated software application. Access to this option requires subscription to the MossWinn Services. For further details click here.
- - to copy the currently displayed spectrum to the clipboard as image. The resolution of the image can be set via the Printer Setup Dialog with selected as printer.
- - to copy the currently displayed spectrum to the clipboard as text, with the velocity values, the measured data, the fit envelope and the subspectra included as comma delimited columns. This option may be used to export numerical data from MossWinn for use in external data processing & graphics software.
- - to copy the currently selected fitted spectrum, the corresponding fit results in two different tabular forms as well as the associated fit report in the form of a single XLS spreadsheet file, in which the spectrum data, the fit result tables and the fit report appear on separate worksheet pages (example).
In order to have the standard deviation of fit parameters included in the XLS file, turn to the Cal StD function before selecting this option. Once created, MossWinn attempts to open the XLS file with an associated software application. Access to this option requires subscription to the MossWinn Services. For further details click here.
- - to copy all the valid insight pages to the clipboard as a single image.
- - to copy the numerical data (including parameter standard errors if calculated) of all the valid insight pages to the clipboard as text. This option may be used to export numerical data from MossWinn for use in external data processing & graphics software.
- - to make a copy of the numerical data of all the valid insight pages in the form of a single XLS spreadsheet file, in which the data of the different insight pages appear on separate worksheets (example).
In order to have standard deviation of fit parameters included in the XLS file, turn to the Cal StD function before selecting this option. Once created, MossWinn attempts to open the XLS file with an associated software application. Access to this option requires subscription to the MossWinn Services. For further details click here.
- - to copy a selected insight page to the clipboard as a single image.
- - to copy the numerical data of a selected insight page to the clipboard as text. This option may be used to export numerical data from MossWinn for use in external data processing & graphics software.
- - to make a copy of the numerical data of the selected insight page in the form of an XLS spreadsheet file (example).
In order to have standard deviation of fit parameters included in the XLS file, turn to the Cal StD function before selecting this option. Once created, MossWinn attempts to open the XLS file with an associated software application. Access to this option requires subscription to the MossWinn Services. For further details click here.
- - to copy the results concerning the currently displayed spectrum to the clipboard in the fit report text format (example).
- - to copy the fit results of all the simultaneously fitted spectra to the clipboard in the fit report text format. The spectra will follow each other according to the spectrum order set previously via the spectrum number popup box.
- - to copy the numerical results concerning all of the simultaneously fitted spectra into the clipboard as comma delimited numerical data in rows, each of which corresponds to a single spectrum. This option may be used to export numerical data from MossWinn for use in external data processing & graphics software.
Concerning the format of the standard deviation values, one of the following options can be chosen:
- with StD values in separate columns
- with StD values in parentheses after values
- with StD values in parentheses as error in last digit(s)
- - to copy the numerical results concerning all of the simultaneously fitted spectra into the clipboard as comma delimited numerical data in columns, each of which corresponds to a single spectrum. This option may be used to export numerical data from MossWinn for use in external data processing & graphics software.
Concerning the format of the standard deviation values, one of the following options can be chosen:
- with StD values in separate rows
- with StD values in parentheses after values
- with StD values in parentheses as error in last digit(s)
- - to copy the correlation matrix of the fit to the clipboard. The following options are available:
- As image (for all spectra)
- As image (for current spectrum)
- As image (value limited, for all spectra)
- As text (for all spectra)
- As text (for current spectrum)
- As text (value limited, for all spectra)
- GUI - The GUI (graphical user interface) menu box provides the possibility to change MossWinn's internal screen resolution according to the following options.
- - to set the internal MossWinn screen resolution to 800 x 400.
- - to set the internal MossWinn screen resolution to 800 x 450.
- - to set the internal MossWinn screen resolution to 800 x 500.
- - to set the internal MossWinn screen resolution to 800 x 650.
- - to set the internal MossWinn screen resolution to 900 x 450.
- - to set the internal MossWinn screen resolution to 900 x 500.
- - to set the internal MossWinn screen resolution to 900 x 650.
- - to set the internal MossWinn screen resolution equal to a value that is optimum for full screen mode by considering the full resolution of the current screen.
- - when selected, the internal MossWinn screen resolution is automatically set to be equal to a value that is optimum for full screen mode whenever the latter is entered by pressing F4.
When full screen mode is entered with this option being unselected, the program sets the internal resolution to the one last used in full screen mode.
- - to set the internal MossWinn screen resolution to 960 x 540, which is considered to be optimum in full screen mode on a FHD (1920 x 1080) screen.
- - to set the internal MossWinn screen resolution to 960 x 600, which is considered to be optimum in full screen mode on a WUXGA (1920 x 1200) screen.
- - to set the internal MossWinn screen resolution to 1280 x 540, which is considered to be optimum in full screen mode on an UW-UXGA (2560 x 1080) screen.
- - to set the internal MossWinn screen resolution to 1280 x 720, which is considered to be optimum in full screen mode on QHD (2560 x 1440) and 4K HD (3840 x 2160) screens.
- - to set the internal MossWinn screen resolution to 1720 x 720, which is considered to be optimum in full screen mode on an UW-QHD (3440 x 1440) screen.
- - to set the internal MossWinn screen resolution to 1920 x 1080, which is considered as a higher resolution alternative on FHD (1920 x 1080) and 4K HD (3840 x 2160) screens.
- - to set the internal MossWinn screen resolution to its maximum value considering the full resolution of the current screen. This option is available only when the program is in full screen mode.
- Coupled W. - Put a check on this box in order to make all the lines of a linear model to have the same line width. Clear the check box in order to set up independent line width parameters (2 in the case of doublets, 3 in the case of sextets) determining the line widths of the lines.
This option is available only for doublet and sextet linear models of the nuclides 57Fe, 119Sn and 125Te. The check box can be invoked via the option of the Contents tab that is accessible in high screen resolution mode.
- Texture - Put a check on this box in order to lift powder constraints of a linear (doublet or sextet) powder geometry model by inserting one additional amplitude type parameter into the model of the selected subspectrum. Clear the check box to return to the constraints of powder geometry.
This option is available only for doublet and sextet linear powder geometry models of the nuclides 57Fe, 119Sn and 125Te. The check box can be invoked via the option of the Contents tab that is accessible in high screen resolution mode.
- Enable neg. amplitudes - Put a check on this box in order to enable amplitude parameters to take on negative values. Normally, amplitude type parameters are forced to be non-negative.
- Contents - click on the Contents tab in order to set the contents of the extra screen area that becomes available on the Model page in high screen resolution modes. Depending on the size of the screen area in question, one or more of the following elements can be made to be displayed.
The selected elements are displayed from left to right in the above order. When the available horizontal space is not sufficient to display more elements, the remaining selected elements become skipped.
The possible functions of the insight pages are listed here.
- Calibration - click on the Calibration tab in order to access the calibration page that allows the calibration of the velocity axis of spectra with known position type parameters (mainly the isomer shift, the hyperfine magnetic field and/or the quadrupole splitting).

- Source - Source nuclide that was used to measure the calibration spectrum. In the present version only 57Fe Mossbauer spectra can be used to calibrate the velocity axis.
- Matrix - Source matrix that was used to measure the calibration spectrum. Built in matrices include
- - with 57Fe isomer shift of +0.114 mm/s relative to α-iron.
- - with 57Fe isomer shift of –0.154 mm/s relative to α-iron.
- - with 57Fe isomer shift of +0.177 mm/s relative to α-iron.
Turn to the menu option in order to define further - single line - matrices by declaring their name and isomer shift relative to α-iron.
- Absorber - Absorber material with known position type parameters, whose 57Fe Mossbauer spectrum was measured in order to calibrate the velocity axis. Absorption materials built in for calibration purposes include
- - α-Fe with 57Fe isomer shift of 0.0 mm/s and hyperfine magnetic field of 33 T.
- - α-Fe measured together with stainless steel.
- - α-Fe measured together with sodium nitroprusside (SNP).
- - α-Fe measured together with SNP and SS (resulting in 1 sextet + 1 doublet + 1 singlet).
- - SNP measured together with SS.
- - sodium nitroprusside (SNP) with 57Fe isomer shift of –0.26 mm/s relative to α-Fe and quadrupole splitting of +1.7034 mm/s.
- - stainless steel (SS) whose 57Fe isomer shift is –0.09 mm/s relative to α-Fe.
Note that during calibration fitting the fixed isomer shift values are always meant relative to the source matrix.
In order to define further absorber fit models for the sake of velocity scale calibration, set the appropriate fit model (on the Model tab) with fixed position type parameters including isomer shift values relative to α-iron, and then by pressing on the Absorber popup on the Calibration tab turn to the menu option .
- Isomer shift reference - Press on this popup box in order to set the reference material for the isomer shift. This setting is taken into account when the calibration fit is accepted. Spectra - calibrated on the basis of the fitted calibration spectrum - when fitted will display isomer shift values relative to the reference material selected here.
If the calibration spectrum is used to calibrate the velocity axis of 57Fe Mossbauer spectra, then is the recommended reference material. When the 57Fe calibration spectrum is used to calibrate the velocity axis of spectra measured with a source nuclide other than 57Fe, then is the recommended reference. In the latter case the velocity axis will be calibrated with absolute velocity values, i.e. the velocity value attributed to a particular channel will be equal to the actual velocity the source was moving with relative to the absorber when the counts in the channel in question were collected. Spectra calibrated in this way will display isomer shift values relative to that of the source material (e.g. 151SmF3 if such a source was used to measure a 151Eu Mossbauer spectrum).
- Velocity waveform - Put a check on the velocity waveform that was used to measure the calibration spectrum. Triangle and Sinusoid refer to the two most often applied waveforms. In the case of folded calibration spectra the Triangle waveform is equivalent to the usual linear velocity scale.
- Folded / Unfolded - Put a check on if the calibration spectrum is a folded spectrum. MossWinn is able to calibrate and fit unfolded spectra as well. In such a case the unfolded calibration spectrum has to be used to calibrate the velocity axis. Put a check on in order to calibrate the velocity axis of an unfolded calibration spectrum (e.g. 2×6 peaks of α-iron).
- Sign of first-channel velocity - Put a check on if the first data count in the data file associated with the calibration spectrum can be attributed to negative source velocity. Otherwise, put a check on . An invalid setting here will result either in an unsatisfactory fit, or in an unrealistic high (considerably higher than 1.0) absolute value of the parameter.
- Calibration mode Active / Inactive - Put a check on in order to let the calibration settings determine the velocity axis and start calibration fitting. Put a check on in order to end calibration mode and return to normal mode when the settings on the calibration tab no longer influence the velocity axis. Normally, after selecting the appropriate options on the calibration tab, one would press on and then perform the calibration fitting. Once an acceptable fit is achieved, one would then press on and leave the fit menu without pressing on .
When calibration mode is active, a new parameter group (with white navigation shapes and value adjusting bars) appears before that of the background - this is the parameter group of the velocity axis that includes the following parameters:
- - The maximum speed of the source during its movement.
- - Electronic delay - measured in channels - of the counting process relative to the movement of the source. Normally its value does not exceed a few channels.
- - The number of channels the whole velocity axis is divided into.
- Calibration constants - This list box displays information on the isomer shift of the source matrix and the reference material.
- Spectrum - Press on the Spectrum tab to have the fitted spectrum displayed with the info line.
- Residual - Press on the Residual tab to have the fitted spectrum and the residual displayed in separate windows (example).
- Insight A, B, C, D, E - Press on one of the Insight tabs in order to have the corresponding insight page displayed. The insight pages all have identical functionalities. By the help of an insight page one can
- display one of the fitted spectra,
- display distribution curves (example),
- display dependence of a fit parameter (e.g. hyperfine magnetic field) on another one (e.g. temperature) in the case of simultaneous fitting of several spectra (example).
Press on the popup boxes following the and labels in order to change the content of an insight page.
- All Spectra - Press on this tab in order to display all of the simultaneously fitted spectra on a single page (example).
The order of spectra is determined by the spectrum order set previously via the spectrum number popup box. Press on any of the spectra with the right mouse button in order to invoke a popup menu with the following menu items.
- - Press on the topmost menu item — displaying the path and the name of the spectrum file — in order to make the spectrum in question to be the selected one whose parameter list is displayed on the parameters panel.
The graph of the currently selected spectrum is displayed with a lightblue background on this page, in contrast with the rest of the spectra whose graph is displayed with a white background.
- - Select this option in order to open the file of the clicked spectrum with the default text editor program.
- - Select this option in order to open the HTML FitLog file associated with the clicked spectrum.
- - Select this option in order to perform a fit by adjusting only those model parameters that belong exclusively to the clicked spectrum.
- - Select this option to copy the image of the clicked spectrum to the clipboard.
- - Select this option to copy the image of the clicked spectrum and that of the associated free-form distribution curves (if any) to the clipboard.
- - Select this option to copy the fit report of the clicked spectrum to the clipboard according to the current state of the fit.
- - Select this option to print the image of the clicked spectrum either to the default printer, or to the one selected here as one of the submenu items.
- - Select this option to print the image of the clicked spectrum and that of the associated free-form distribution curves (if any) either to the default printer, or to the one selected here as one of the submenu items.
- - Select this option to print the fit report of the clicked spectrum (according to the current state of the fit) either to the default printer, or to the one selected here as one of the submenu items.
- - Select this option to remove the clicked spectrum from the (simultaneous) fit.
- Insights - Press on this tab in order to display all of the insight pages (Insights A-F) on a single page (example).
Press on any of the insight pages with the right mouse button in order to invoke a popup menu with the following menu items.
- - The topmost menu item displays the name of the clicked insight page.
- - Select to connect/disconnect the data points on the clicked insight page.
- - Select this option in order to enable/disable the display of error bars on the clicked insight page. Note that error bars are displayed only when the standard deviation of the parameters were previously calculated via the Cal StD menu box.
When shown, the error bars extend 1×σ (standard deviation) both in the positive and the negative direction from the associated data point. When the standard deviation in question is too low compared to the corresponding range of the graph, the error bars may be to small to be visible.
- - Select the color of error bars from the appearing submenu.
- - Select this option to copy the numerical data (including the errors if any) of the clicked insight page to the clipboard as text. This option may be used to export numerical data from MossWinn for use in external data processing & graphics software.
- - Select this option to copy the image of the clicked insight page to the clipboard.
- - Select this option to copy all the valid insight pages to the clipboard as a single image.
- - Select this option to print the image of the clicked insight page either to the default printer, or to the one selected here as one of the submenu items.
- - Select this option to print the image of all the valid insight pages either to the default printer, or to the one selected here as one of the submenu items.
- Info line - Press on the info line popup box (displaying Alpha iron above) in order to invoke a popup menu allowing one to determine the information displayed by the info line, to set attributes of spectrum graphics, and to open the HTML FitLog file created previously for the current spectrum. The FitLog file (if exists) is opened in the default web browser application.
- The goodness of the (single) fit is displayed in the bottom of the spectrum panel in the form of
Chi: () Goodness:
If the chosen theoretical model is correct, then an acceptable fit should provide a normalized chisquare close to 1, and a goodness of fit value higher than about 0.001 .
In the case of simultaneous fitting of several spectra, these values characterize only the fit of the current spectrum, as if it was the only one that is fitted. In contrast, the corresponding values displayed at the bottom of the parameters' panel characterize the simultaneous fit as a whole.
|

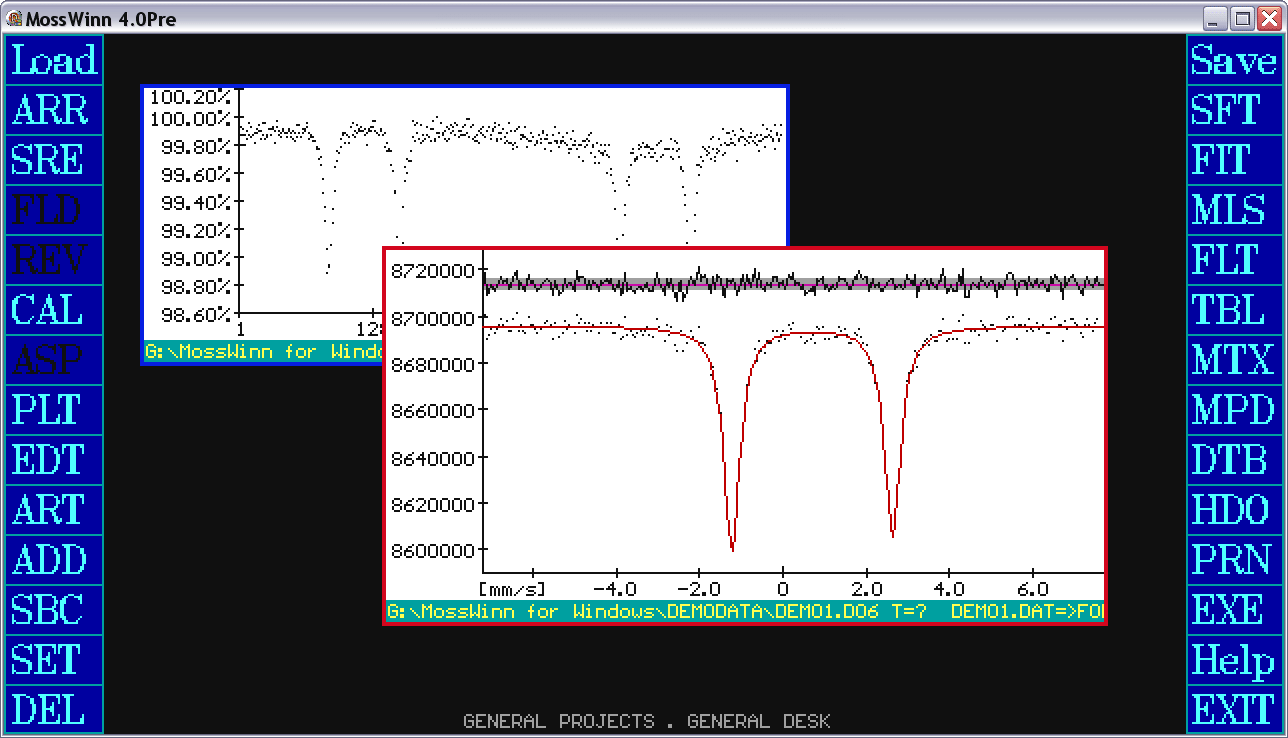



 and its temporary copy (bottom).")




 menu")


 and after (bottom) applying the reverse operation to a 151Eu Mossbauer spectrum.")
 and after (bottom) applying the reverse operation to a 151Eu Mossbauer spectrum.")
.")
.")













 with its normalized version (bottom).")
 and its stripped version (bottom).")
 and the spectrum after horizontal reflection (bottom).")
 and their convolution with a Gaussian of unit area and FWHM of 2 mm/s (bottom).")
 and their convolution with a Lorentzian of unit amplitude and FWHM of 1 mm/s (bottom).")
 and after (bottom) the addition of normally distributed statistical noise.")

 and after (bottom) adding up neighboring channels of a spectrum.")
 was subtracted from the original (left-top) giving rise to the blue-framed result (right-top).")
 and the corresponding residual (bottom).")


 resolution.")




 menu")
 menu")
 menu")
 menu")




 menu")

 menu")



 menu list")

")



")





















 options - screen resolution")

























 menu")










{kind=link}
{kind=link}
{kind=link}
{kind=link}
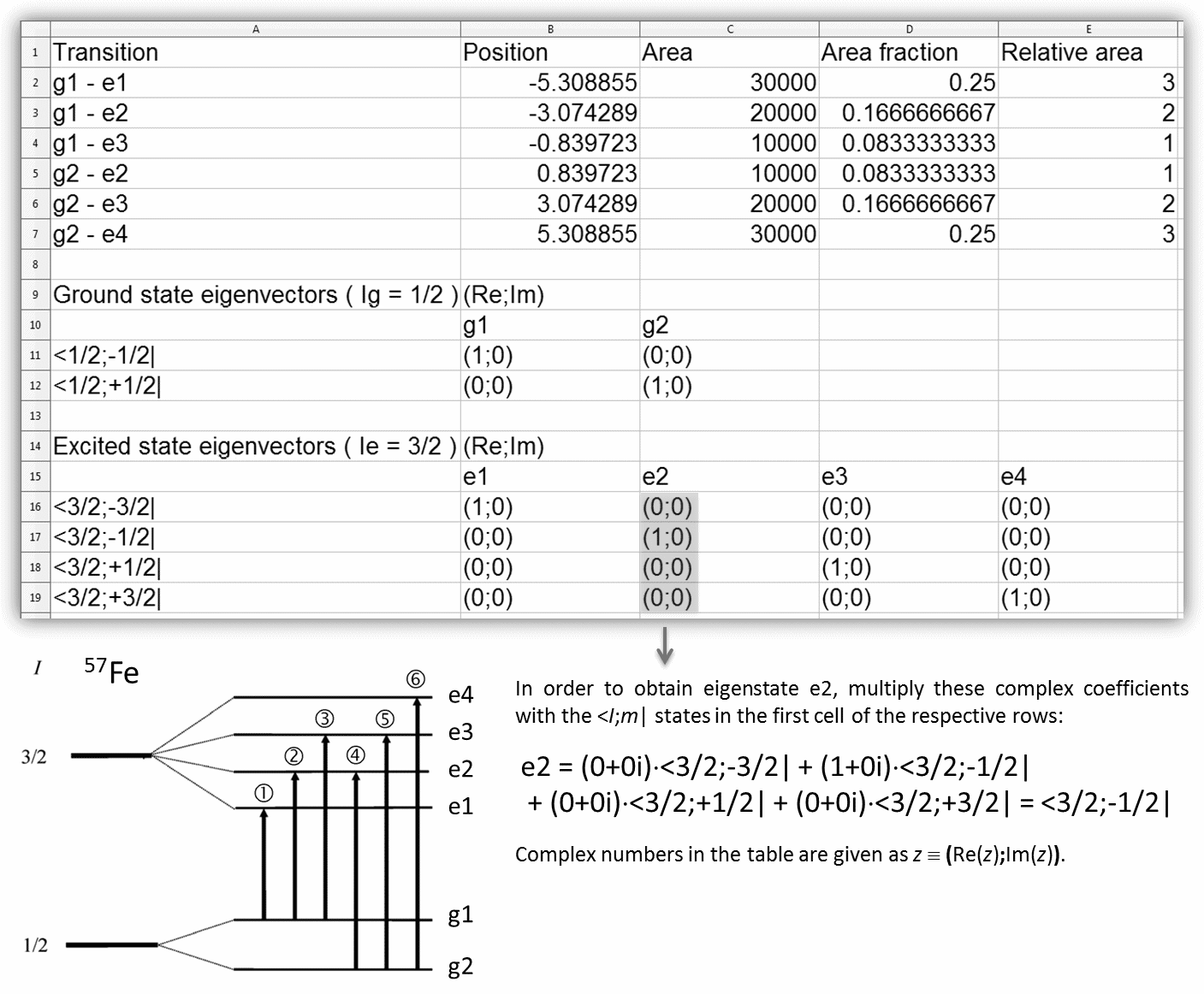{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}